Integration of scRNA and multi-omic datasets
[1]:
import numpy as np
import anndata as ad
import scanpy as sc
import scvelo as scv
import torch
from sklearn.decomposition import PCA
import umap
import os
import multivelovae as vv
from datetime import datetime
[2]:
torch.cuda.is_available()
[2]:
True
[3]:
now = datetime.now()
date = now.strftime("%m_%d_%Y")
Read in processed data and define places to store output
[4]:
adata = sc.read_h5ad('3423-MV-2-8489-MV-1-GSM5460410_adata_postpro_concat.h5ad')
adata_atac = sc.read_h5ad('3423-MV-2-8489-MV-1-GSM5460410_adata_atac_postpro_concat.h5ad')
[5]:
gene_plot = ['AZU1', 'HBB', 'HDC', 'LYZ', 'PF4', 'SPINK2']
np.isin(gene_plot, adata.var_names)
[5]:
array([ True, True, True, True, True, True])
[6]:
'PROM1' in adata.var_names
[6]:
True
[7]:
dataset = "3423-MV-2-8489-MV-1-GSM5460410"
model_path_base = f"checkpoints/{dataset}"
figure_path_base = f"figures/{dataset}"
data_path_base = f"data/{dataset}"
[8]:
adata, adata_atac
[8]:
(AnnData object with n_obs × n_vars = 27841 × 1044
obs: 'total_unspliced', 'total_spliced', 'log1p_total_unspliced', 'log1p_total_spliced', 'fraction_u', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_20_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_rb', 'log1p_total_counts_rb', 'pct_counts_rb', 'total_counts_hla', 'log1p_total_counts_hla', 'pct_counts_hla', 'total_counts_hb', 'log1p_total_counts_hb', 'pct_counts_hb', 'outlier', 'initial_size_unspliced', 'initial_size_spliced', 'initial_size', 'n_counts', 'S_score', 'G2M_score', 'phase', 'CC_difference', 'total_unspliced2', 'total_spliced2', 'log1p_total_unspliced2', 'log1p_total_spliced2', 'fraction_u2', 'n_c', 'n_Mu', 'n_Ms', 'leiden', 'total_chromatin', 'total_unspliced3', 'total_spliced3', 'log1p_total_unspliced3', 'log1p_total_spliced3', 'fraction_u3', 'total_Mc', 'total_Mu', 'total_Ms', 'batch', 'n_Mc'
var: 'gene_ids', 'feature_types', 'mt', 'rb', 'hla', 'hb', 'HVG-Multiome 1', 'n_cells-Multiome 1', 'n_cells_by_counts-Multiome 1', 'mean_counts-Multiome 1', 'log1p_mean_counts-Multiome 1', 'pct_dropout_by_counts-Multiome 1', 'total_counts-Multiome 1', 'log1p_total_counts-Multiome 1', 'gene_count_corr-Multiome 1', 'highly_variable-Multiome 1', 'mean-Multiome 1', 'std-Multiome 1', 'HVG-Multiome 2', 'n_cells-Multiome 2', 'n_cells_by_counts-Multiome 2', 'mean_counts-Multiome 2', 'log1p_mean_counts-Multiome 2', 'pct_dropout_by_counts-Multiome 2', 'total_counts-Multiome 2', 'log1p_total_counts-Multiome 2', 'gene_count_corr-Multiome 2', 'highly_variable-Multiome 2', 'mean-Multiome 2', 'std-Multiome 2', 'HVG-scRNA', 'n_cells-scRNA', 'n_cells_by_counts-scRNA', 'mean_counts-scRNA', 'log1p_mean_counts-scRNA', 'pct_dropout_by_counts-scRNA', 'total_counts-scRNA', 'log1p_total_counts-scRNA', 'gene_count_corr-scRNA', 'highly_variable-scRNA', 'mean-scRNA', 'std-scRNA'
uns: 'batch_colors', 'leiden_colors', 'neighbors', 'umap'
obsm: 'X_pca', 'X_umap'
layers: 'Ms', 'Mu', 'spliced', 'unspliced'
obsp: 'connectivities', 'distances',
AnnData object with n_obs × n_vars = 27841 × 1044
obs: 'n_counts', 'batch'
layers: 'Mc')
[9]:
adata.obs['leiden'].cat.categories
[9]:
Index(['CMP', 'DC', 'Erythrocyte', 'GMP', 'Granulocyte', 'HSC', 'LMPP', 'MEP',
'Mast Cells', 'Megakaryocyte', 'Platelet', 'Prog B', 'Prog DC',
'Prog MK'],
dtype='object')
[10]:
adata.obs['batch'].cat.categories
[10]:
Index(['Multiome 1', 'Multiome 2', 'scRNA'], dtype='object')
[11]:
os.makedirs(figure_path_base, exist_ok=True)
scv.pl.scatter(adata, basis='umap', color='leiden', legend_loc='right margin')
scv.pl.scatter(adata, basis='umap', color='batch', legend_loc='right margin')
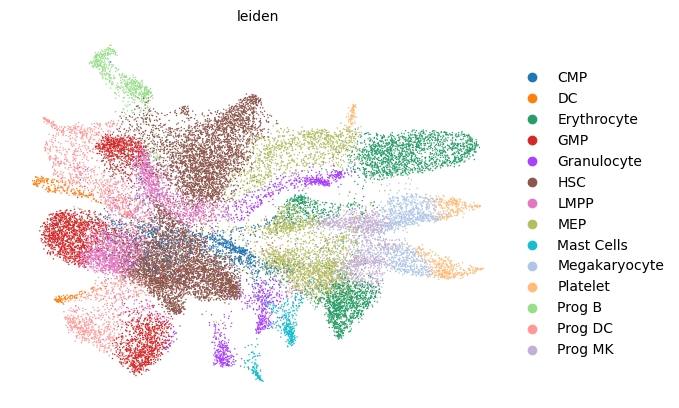
[12]:
figure_path = figure_path_base+'/'+date
model_path = model_path_base+'/'+date
data_path = data_path_base
Initialize and train a MultiVeloVAE
[13]:
key = "vae"
[14]:
torch.manual_seed(2022)
np.random.seed(2022)
model = vv.VAEChrom(adata,
adata_atac,
batch_key='batch',
ref_batch=1,
batch_hvg_key='highly_variable',
var_to_regress=['total_unspliced3'],
device='cuda:0',
rna_only_idx=2,
plot_init=False,
gene_plot=gene_plot,
cluster_key="leiden",
figure_path=figure_path,
embed="umap")
model.train(plot=False,
gene_plot=gene_plot,
figure_path=figure_path,
embed="umap")
model.save_model(model_path)
model.save_anndata(data_path, file_name="out.h5ad")
Running in mixed RNA-only mode.
CVAE enabled. Performing batch effect correction.
Reference batch set to 1 (Multiome 2).
Latent dimension set to 9.
KL weights set to 4.00 4.00.
Found ['total_unspliced3'] in obs. Regressing them out.
Batch 0 sparsity: 0.095
Batch 1 sparsity: 0.108
Batch 2 sparsity: 0.169
Learning rate set to 7.5e-4 based on data sparsity.
Early stop threshold set to 2.6.
Using Gaussian Prior.
Initializing using the steady-state and dynamical models.
956 out of 1044 = 91.6% genes have good ellipse fits.
KS-test result: [1. 1. 1. 1. 1. 1. 1.]
Assigning cluster 5 to repressive.
Initial induction: 809, repression: 235 out of 1044.
Computing scaling factors for each batch class.
-------------------------- Train a MultiVeloVAE -------------------------
********* Creating Training and Validation Datasets *********
Total Number of Iterations Per Epoch: 77, test iteration: 152
********* Finished. *********
********* Stage 1 *********
Epoch 1: Train ELBO = 119.073, Test ELBO = -778723.808 Total Time = 0 h : 0 m : 1 s
Epoch 50: Train ELBO = 1407.613, Test ELBO = 1407.692 Total Time = 0 h : 0 m : 29 s
Epoch 100: Train ELBO = 1428.686, Test ELBO = 1423.719 Total Time = 0 h : 0 m : 58 s
********* Stage 1: Early Stop Triggered at epoch 121. *********
********* Retrieving best model from iteration 9121. *********
********* Stage 2 *********
Cell-wise KNN estimation.
Using 616 latent neighbors to select ancestors.
Percentage of Invalid Sets: 0.000
Average Set Size: 100
Finished. Actual Time: 0 h : 0 m : 24 s
********* Velocity Refinement Round 1 *********
Epoch 127: Train ELBO = 1305.002, Test ELBO = 1308.716 Total Time = 0 h : 1 m : 37 s
********* Round 1: Early Stop Triggered at epoch 127. *********
********* Retrieving best model from iteration 9702. *********
Cell-wise KNN estimation.
Finished. Actual Time: 0 h : 0 m : 3 s
********* Velocity Refinement Round 2 *********
Epoch 131: Train ELBO = 1273.187, Test ELBO = 1277.815 Total Time = 0 h : 1 m : 44 s
********* Round 2: Early Stop Triggered at epoch 131. *********
********* Retrieving best model from iteration 10002. *********
Change in x0: 0.210
Cell-wise KNN estimation.
Finished. Actual Time: 0 h : 0 m : 3 s
********* Velocity Refinement Round 3 *********
Epoch 135: Train ELBO = 1243.057, Test ELBO = 1247.244 Total Time = 0 h : 1 m : 50 s
********* Round 3: Early Stop Triggered at epoch 135. *********
********* Retrieving best model from iteration 10302. *********
Change in x0: 0.163
Cell-wise KNN estimation.
Finished. Actual Time: 0 h : 0 m : 3 s
********* Velocity Refinement Round 4 *********
Epoch 139: Train ELBO = 1217.538, Test ELBO = 1221.508 Total Time = 0 h : 1 m : 57 s
********* Round 4: Early Stop Triggered at epoch 139. *********
********* Retrieving best model from iteration 10602. *********
Change in x0: 0.131
Cell-wise KNN estimation.
Finished. Actual Time: 0 h : 0 m : 3 s
********* Velocity Refinement Round 5 *********
Epoch 143: Train ELBO = 1194.960, Test ELBO = 1199.444 Total Time = 0 h : 2 m : 4 s
********* Round 5: Early Stop Triggered at epoch 143. *********
********* Retrieving best model from iteration 10902. *********
Change in x0: 0.124
Cell-wise KNN estimation.
Finished. Actual Time: 0 h : 0 m : 3 s
********* Velocity Refinement Round 6 *********
Epoch 147: Train ELBO = 1172.169, Test ELBO = 1176.171 Total Time = 0 h : 2 m : 10 s
********* Round 6: Early Stop Triggered at epoch 147. *********
********* Retrieving best model from iteration 11202. *********
Change in x0: 0.121
Final: Train ELBO = 1172.169, Test ELBO = 1176.171
********* Finished. Total Time = 0 h : 2 m : 11 s *********
Using reference batch for latent variable computation.
Computing velocity.
Selected 776 velocity genes.
Writing anndata output to file.
Downstream analyses
[15]:
z = adata.obsm[f'{key}_z']
t = adata.obs[f'{key}_time'].to_numpy()
rho = adata.layers[f'{key}_rho']
kc = adata.layers[f'{key}_kc']
[16]:
# UMAP embedding from latent cell space using reference label
umap_obj = umap.UMAP(n_neighbors=30, n_components=2, min_dist=0.25, random_state=2022)
z_umap = umap_obj.fit_transform(z)
# UMAP embedding from latent cell space using original batch labels
z_batch = adata.obsm[f'{key}_z_batch']
umap_obj = umap.UMAP(n_neighbors=30, n_components=2, min_dist=0.25, random_state=2022)
z_umap2 = umap_obj.fit_transform(z_batch)
[17]:
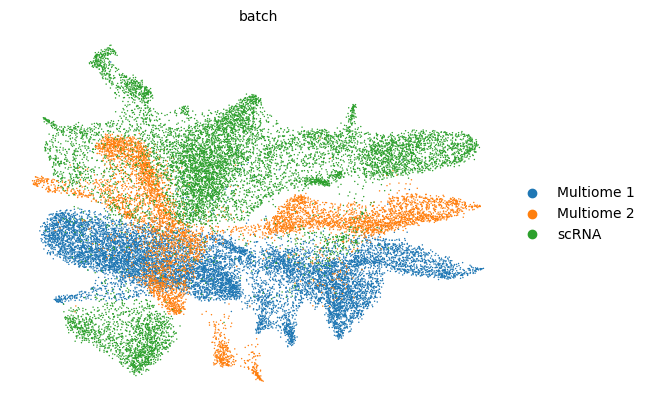
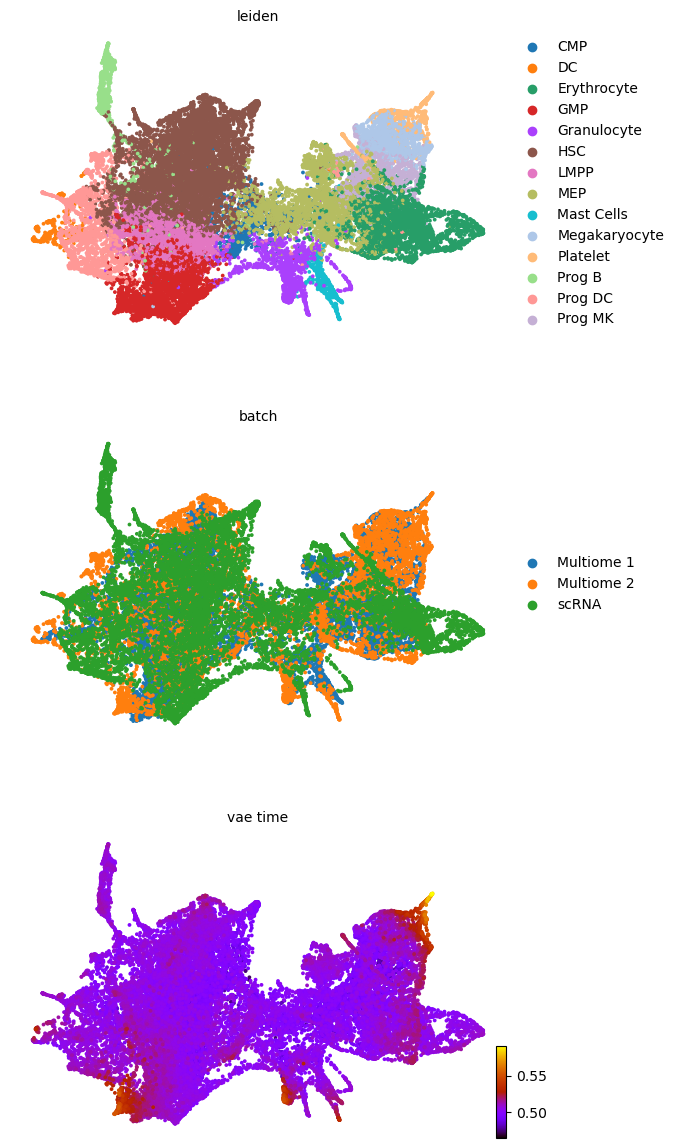
scv.pl.scatter(adata, basis='umap', color=['leiden', 'batch', f'{key}_time'], legend_loc='right', color_map='gnuplot', ncols=1, size=30)
adata.obsm['X_z_umap'] = z_umap
scv.pl.scatter(adata, basis='z_umap', color=['leiden', 'batch', f'{key}_time'], legend_loc='right', color_map='gnuplot', ncols=1, size=30, save=f'{figure_path}/{dataset}_z_batch_time_{key}.png')
adata.obsm['X_z_umap2'] = z_umap2
scv.pl.scatter(adata, basis='z_umap2', color=['leiden', 'batch', f'{key}_time'], legend_loc='right', color_map='gnuplot', ncols=1, size=30, save=f'{figure_path}/{dataset}_z_batch_time2_{key}.png')
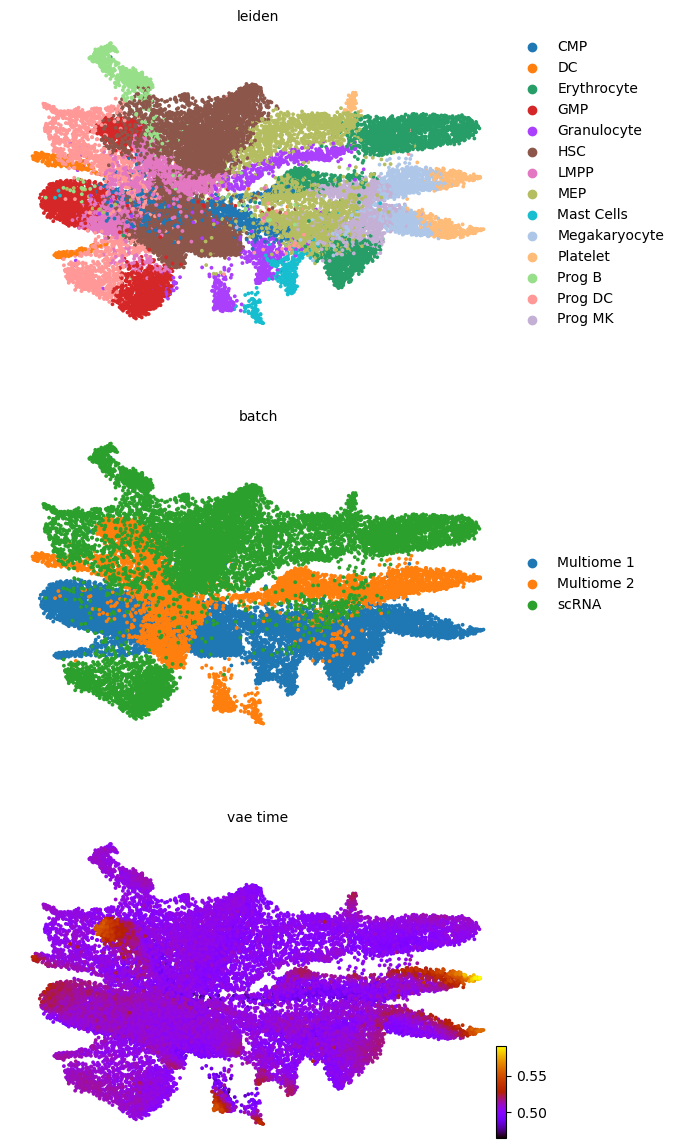
saving figure to file figures/3423-MV-2-8489-MV-1-GSM5460410/06_04_2025/3423-MV-2-8489-MV-1-GSM5460410_z_batch_time_vae.png
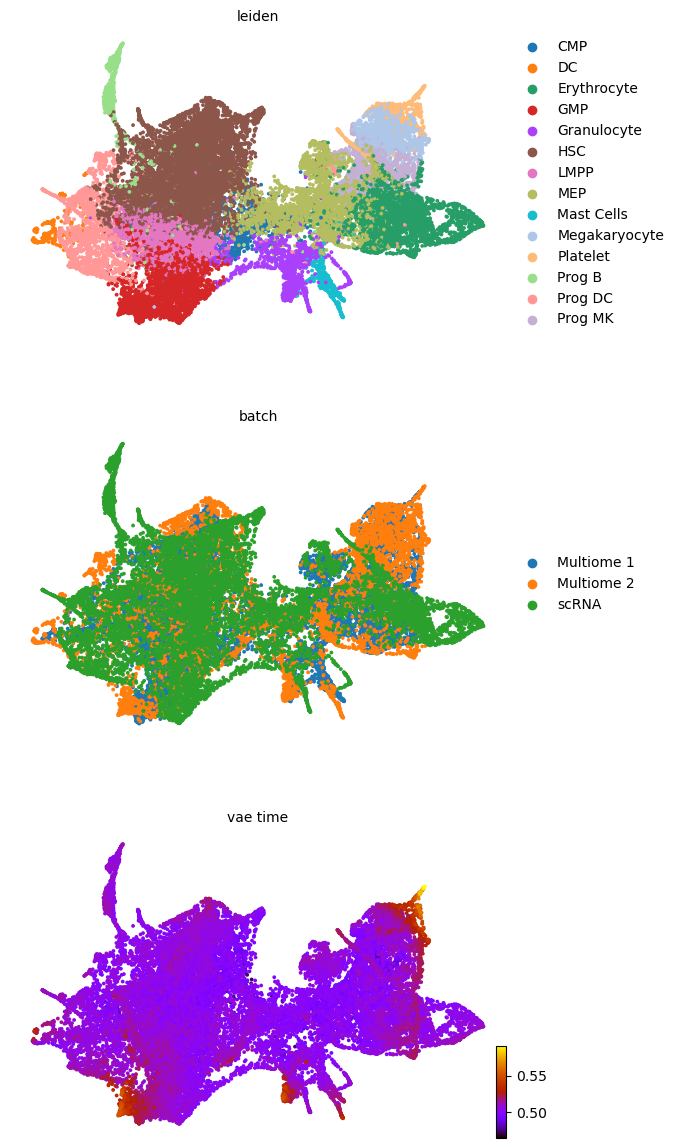
saving figure to file figures/3423-MV-2-8489-MV-1-GSM5460410/06_04_2025/3423-MV-2-8489-MV-1-GSM5460410_z_batch_time2_vae.png
[18]:
adata.obsm[f'{key}_latent'] = np.concatenate((adata.obsm[f'{key}_z'], adata.obsm[f'{key}_t']), axis=1)
sc.pp.neighbors(adata, use_rep=f'{key}_latent', n_neighbors=50)
[19]:
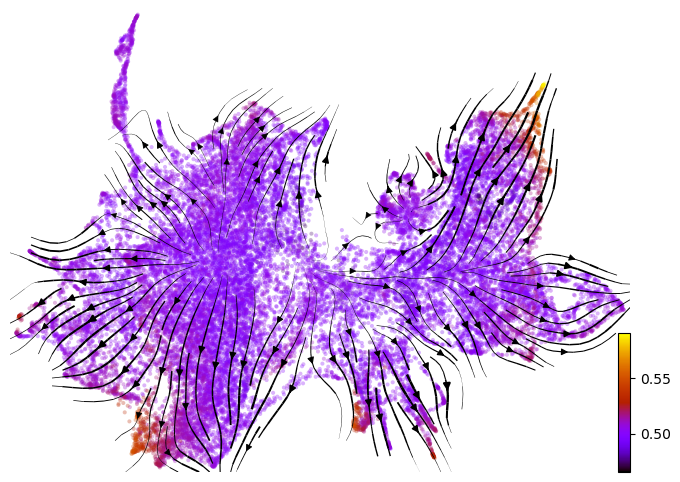
# Compute velocity graph on Z UMAP and visualize it, together with batch labels and inferred latent time
adata.obsm['X_z_umap'] = z_umap
vv.model.velocity_graph(adata, key=key, batch_corrected=True)
vv.velocity_embedding_stream(adata, key=key, basis='z_umap', color='leiden', title="", figsize=(8,6), legend_loc='right margin')
vv.velocity_embedding_stream(adata, key=key, basis='z_umap', color='batch', title="", figsize=(8,6), legend_loc='right margin')
vv.velocity_embedding_stream(adata, key=key, basis='z_umap', color=f'{key}_time', color_map='gnuplot', title="", figsize=(8,6), legend_loc='right margin')
computing velocity graph (using 1/8 cores)
finished (0:01:17) --> added
'vae_velocity_norm_graph', sparse matrix with cosine correlations (adata.uns)
computing velocity embedding
finished (0:00:03) --> added
'vae_velocity_norm_z_umap', embedded velocity vectors (adata.obsm)


[20]:
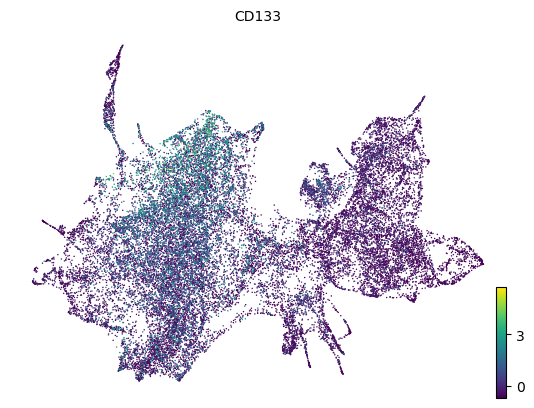
# HSC marker gene CD133 (PROM1)
scv.pl.scatter(adata, color='PROM1', basis='z_umap', title='CD133')
[21]:
adata.obs['clusters'] = adata.obs['leiden']
[22]:
# Subset to scRNA cells only for later use
adata_sub = adata[adata.obs['batch']=='scRNA',:].copy()
adata_atac_sub = adata_atac[adata.obs['batch']=='scRNA',:].copy()
[23]:
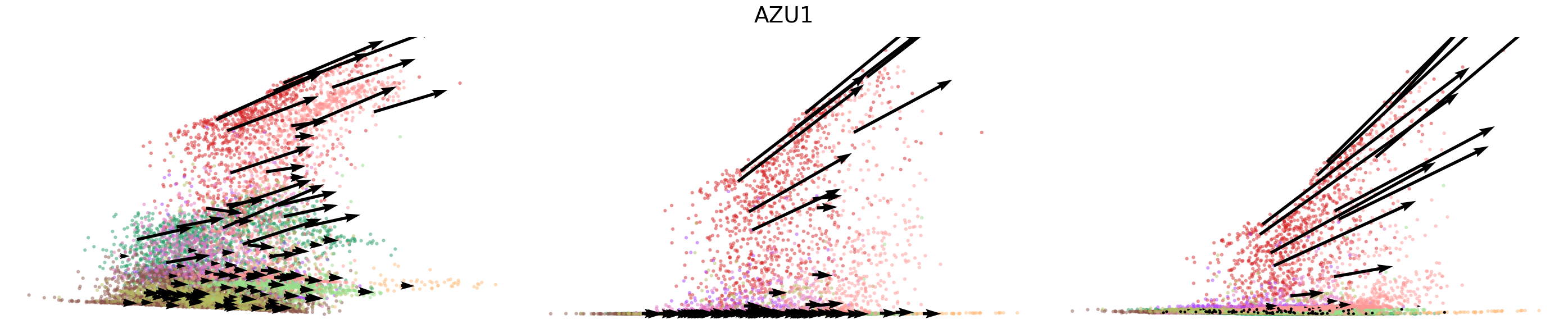
# Dynamics plots for selected genes with velocity arrows, colored by cell types
np.random.seed(2022)
for gene in gene_plot:
vv.plot_vel(adata_sub.obs[f'{key}_time'],
adata_sub[:,gene].layers[f'{key}_chat'],
adata_sub[:,gene].layers[f'{key}_uhat'],
adata_sub[:,gene].layers[f'{key}_shat'],
adata_sub[:,gene].layers[f'{key}_velocity_c'],
adata_sub[:,gene].layers[f'{key}_velocity_u'],
adata_sub[:,gene].layers[f'{key}_velocity'],
cell_labels=adata_sub.obs['leiden'].to_numpy(),
cell_type_colors=model.cell_type_colors,
title=gene,
frame_on=False,
axis_on=False,
legend=False)
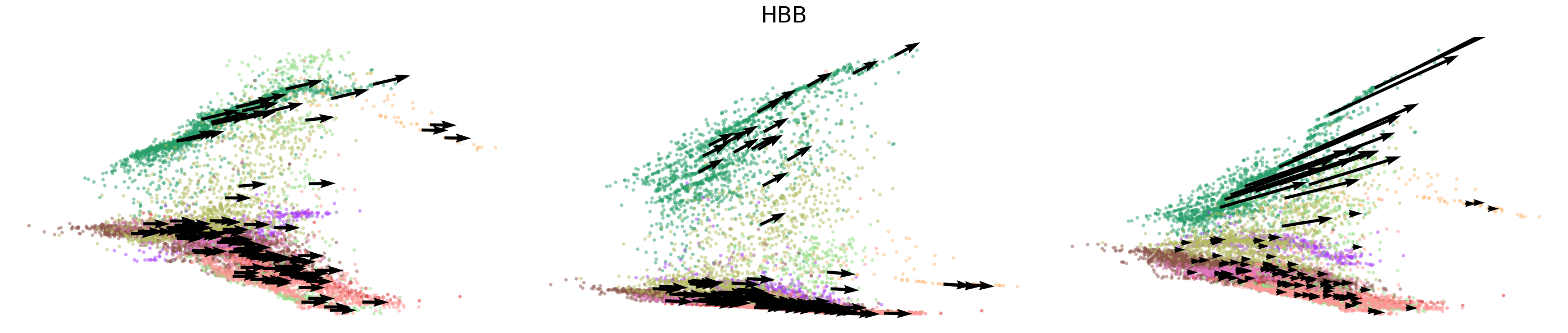
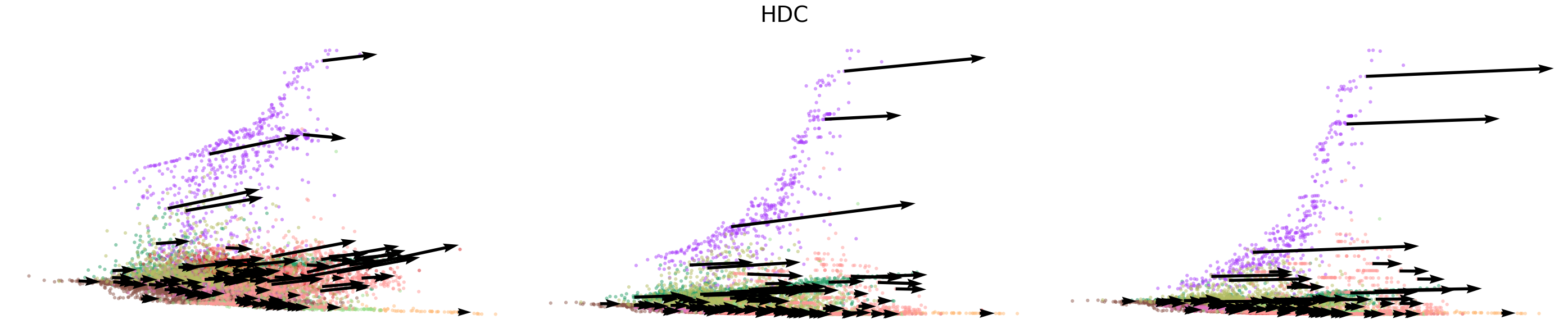
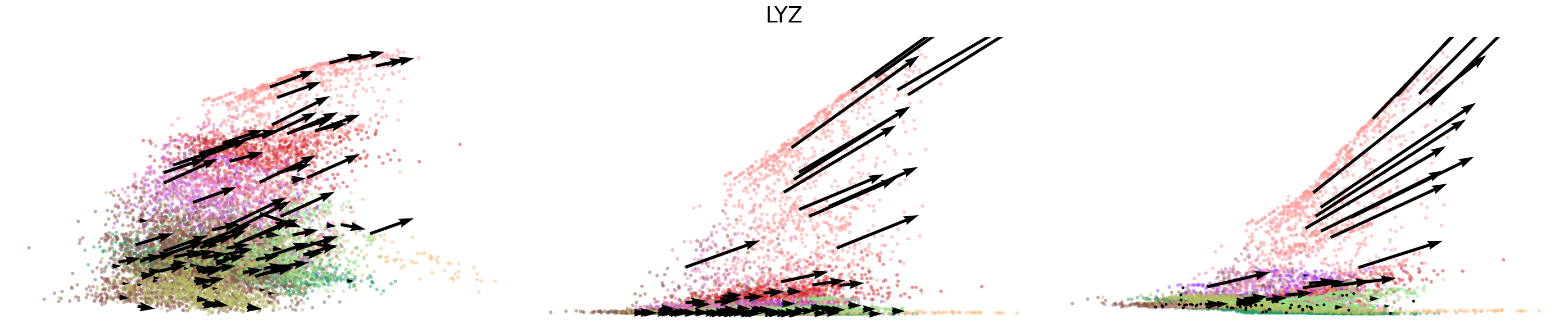
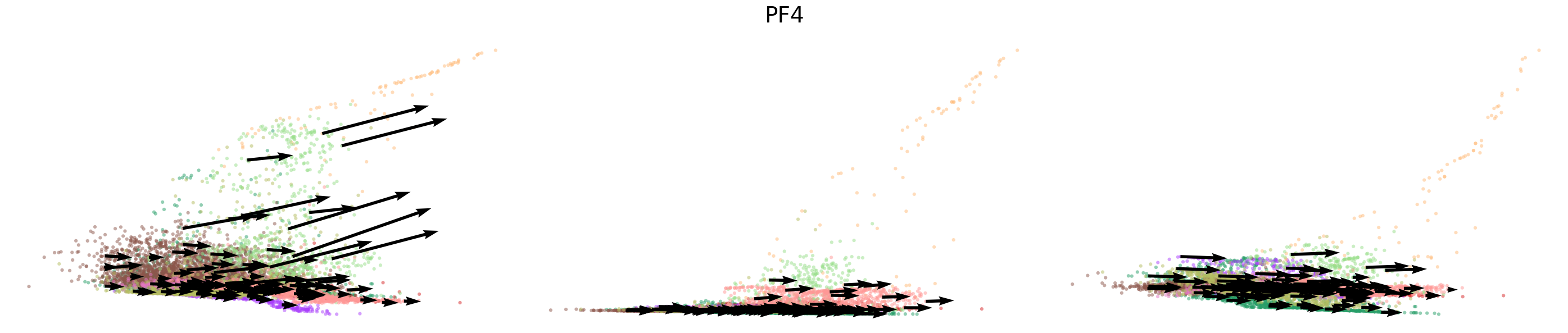
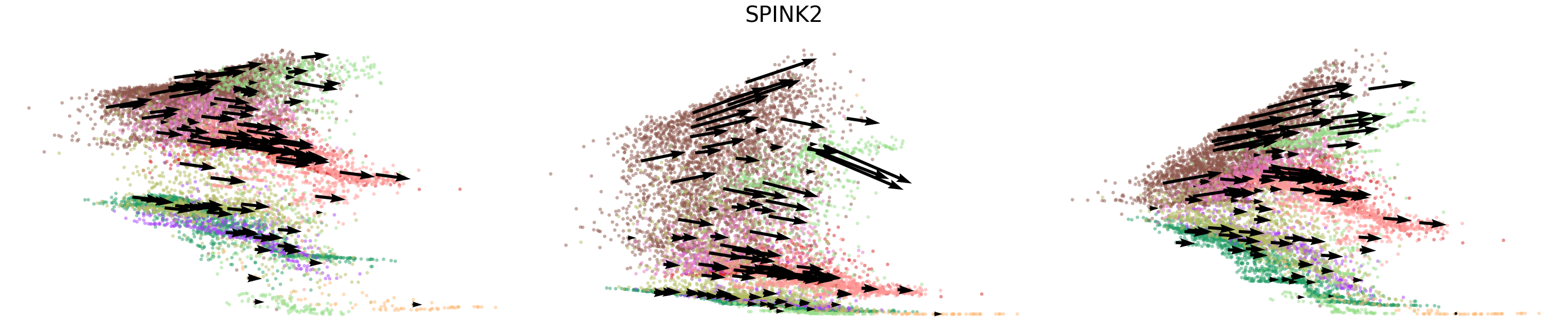
[24]:
# Dynamics plots for selected genes with velocity arrows, colored by batch labels
batch_raw = adata.obs['batch'].cat.categories.to_numpy()
batch_colors = {batch_raw[i]: adata.uns['batch_colors'][i] for i in range(len(batch_raw))}
np.random.seed(2022)
for gene in gene_plot:
vv.plot_vel(adata.obs[f'{key}_time'],
adata[:,gene].layers[f'{key}_chat'],
adata[:,gene].layers[f'{key}_uhat'],
adata[:,gene].layers[f'{key}_shat'],
adata[:,gene].layers[f'{key}_velocity_c'],
adata[:,gene].layers[f'{key}_velocity_u'],
adata[:,gene].layers[f'{key}_velocity'],
cell_labels=adata.obs['batch'].to_numpy(),
cell_type_colors=batch_colors,
title=gene,
frame_on=False,
axis_on=False,
legend=False)
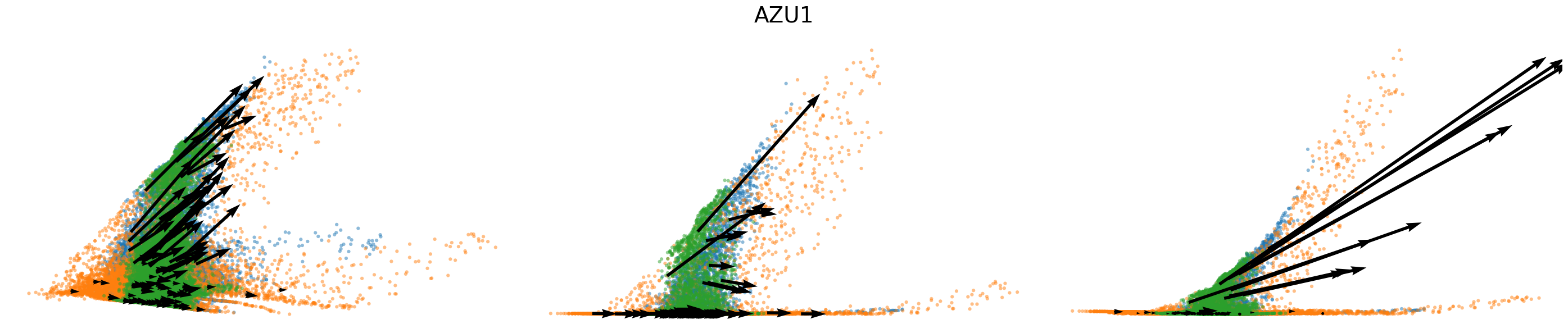
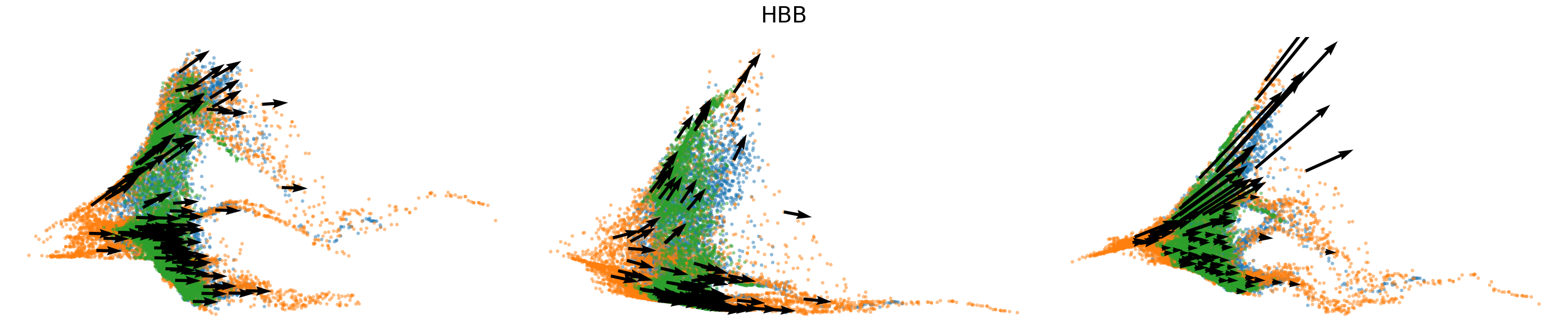
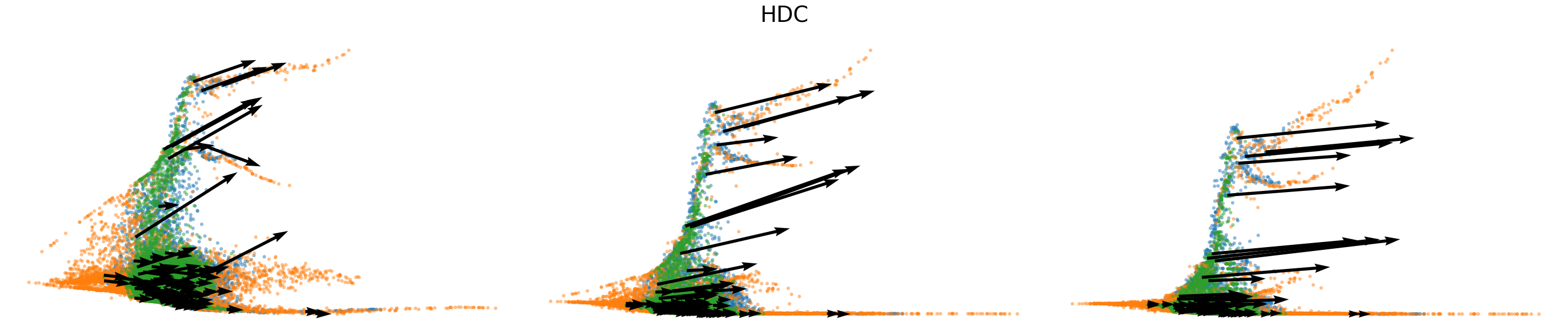
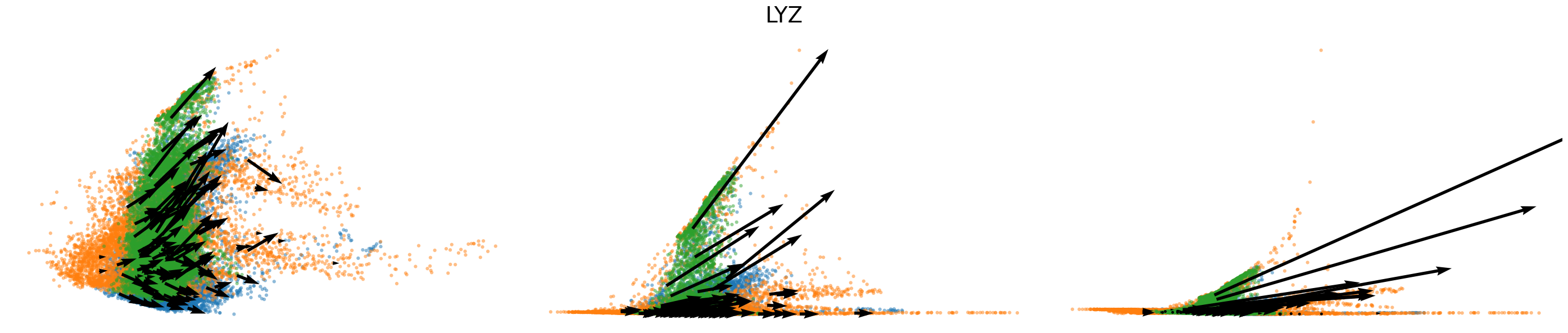
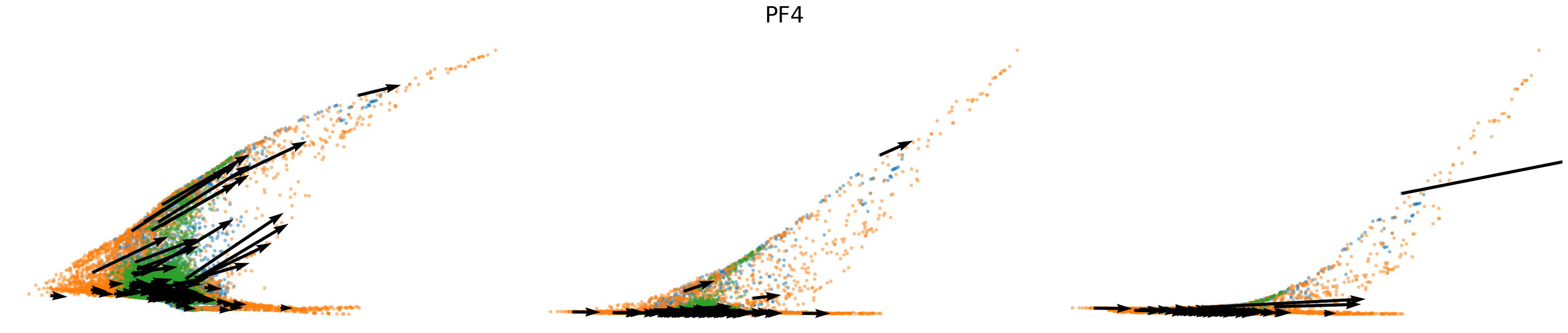

[25]:
# Dynamic plots of modality level by latent time for genes of interest
vv.dynamic_plot(adata, adata_atac, gene_plot, color_by='leiden', batch_correction=True)
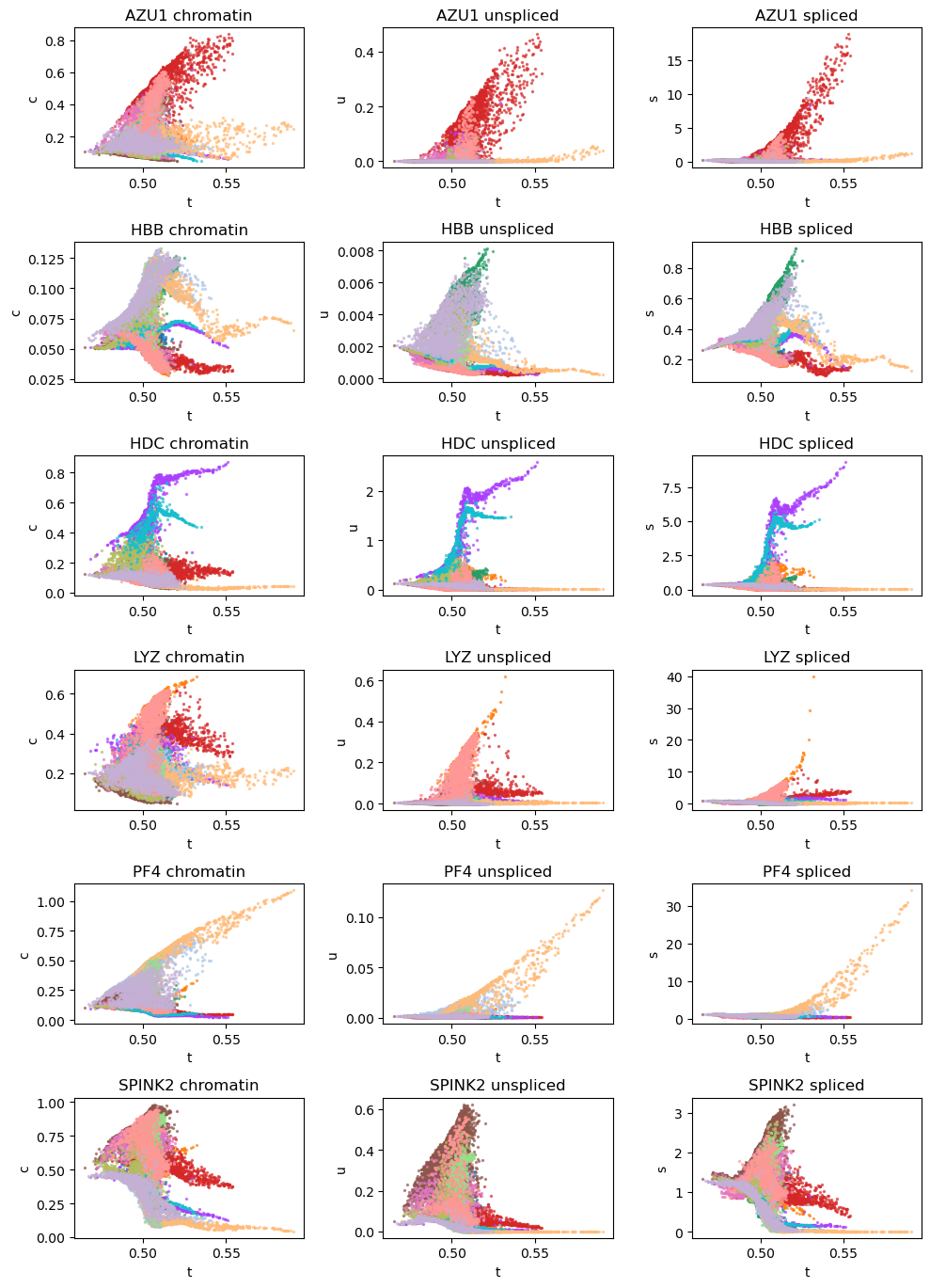
[26]:
# Dynamic plots of modality level by latent time for genes of interest, for scRNA cells only
vv.dynamic_plot(adata_sub, adata_atac_sub, gene_plot, color_by='leiden', batch_correction=True)
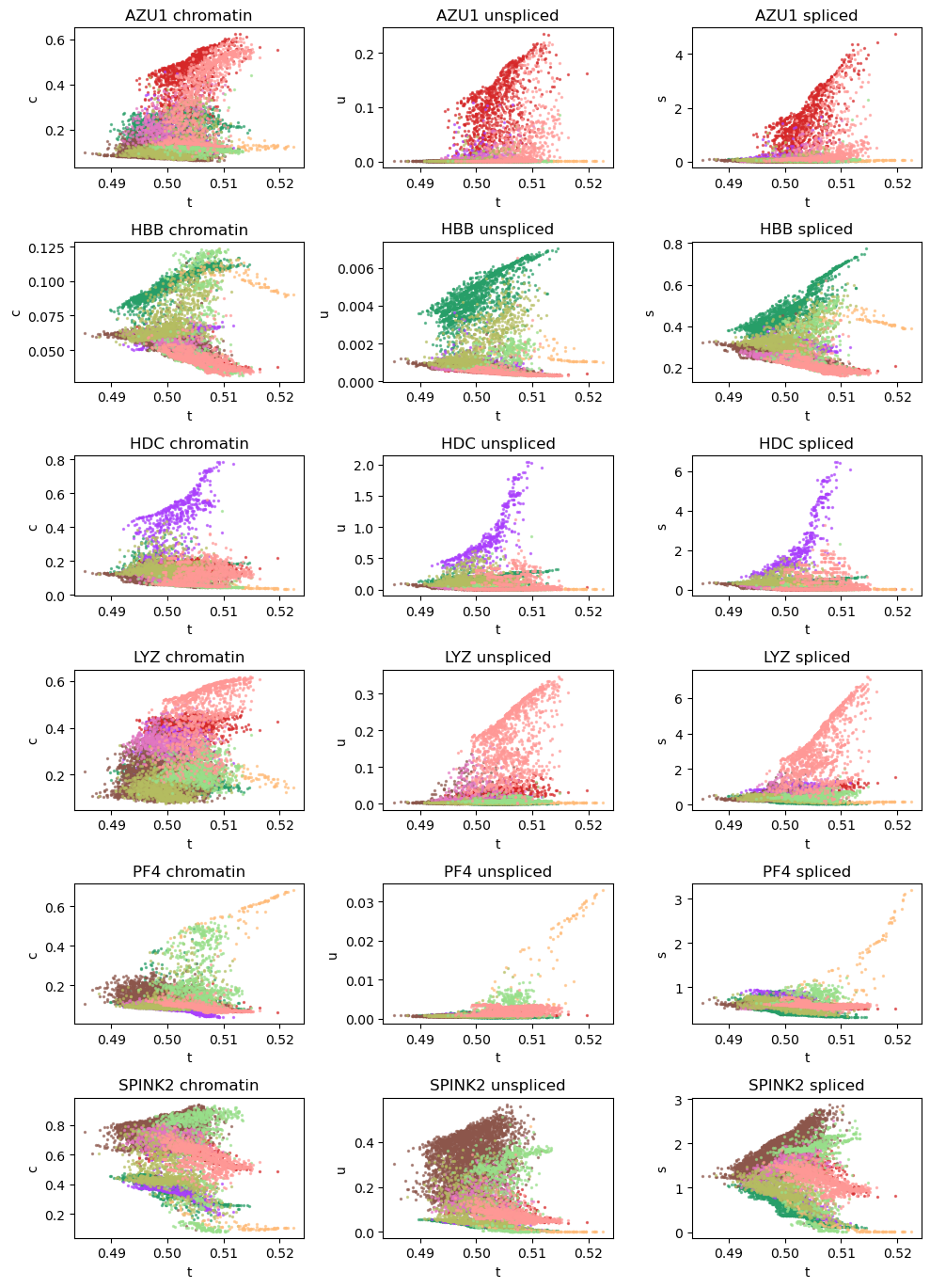
Plot phase portraits of genes with the highest likelihoods
[27]:
top_genes = adata[:, adata.var[f'{key}_velocity_genes']].var[f'{key}_likelihood'].sort_values(ascending=False).index
[28]:
scv.pl.scatter(adata, basis=top_genes[:20], x=f'{key}_uhat', y=f'{key}_chat', color='batch', ncols=5)
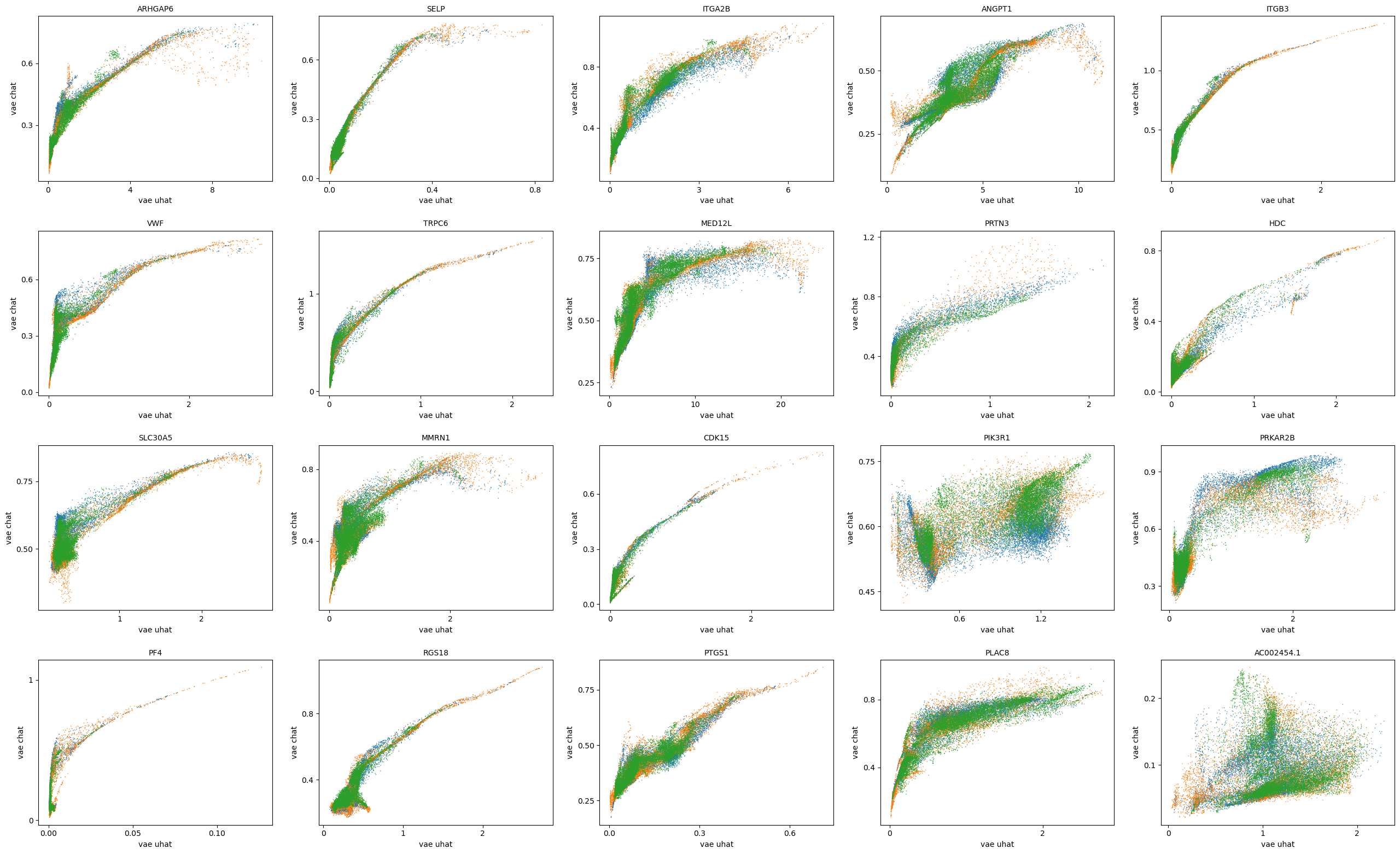
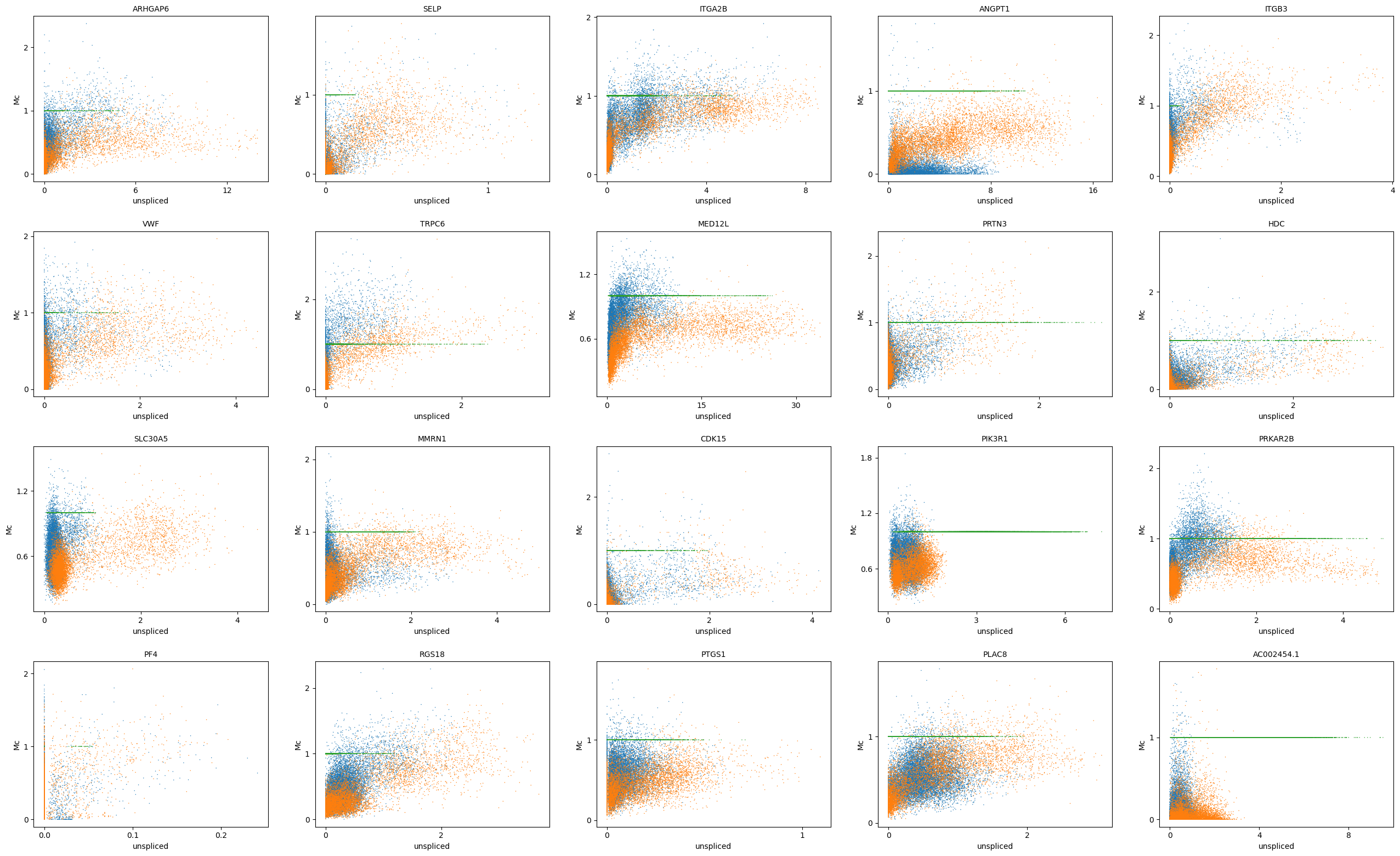
[29]:
adata.layers['Mc'] = adata_atac.layers['Mc']
scv.pl.scatter(adata, basis=top_genes[:20], x='Mu', y='Mc', color='batch', ncols=5)
Plot phase portraits of selected marker genes
[30]:
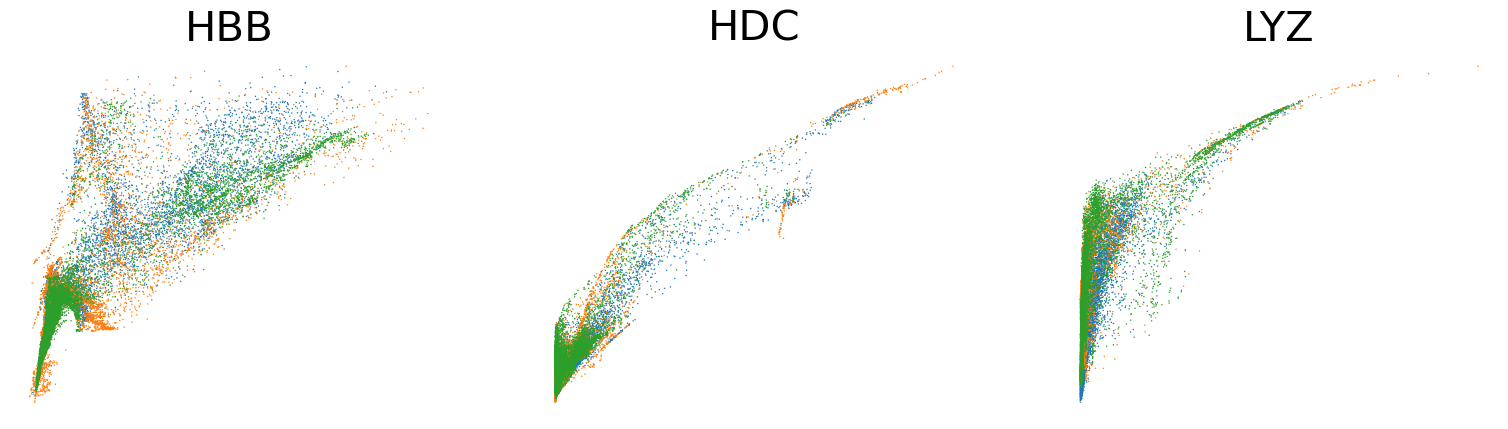
scv.pl.scatter(adata, basis=['HBB', 'HDC', 'LYZ'], x='Mu', y='Mc', color='leiden', ncols=3, frameon=False, fontsize=30)
scv.pl.scatter(adata, basis=['HBB', 'HDC', 'LYZ'], x=f'{key}_uhat', y=f'{key}_chat', color='leiden', ncols=3, frameon=False, fontsize=30)
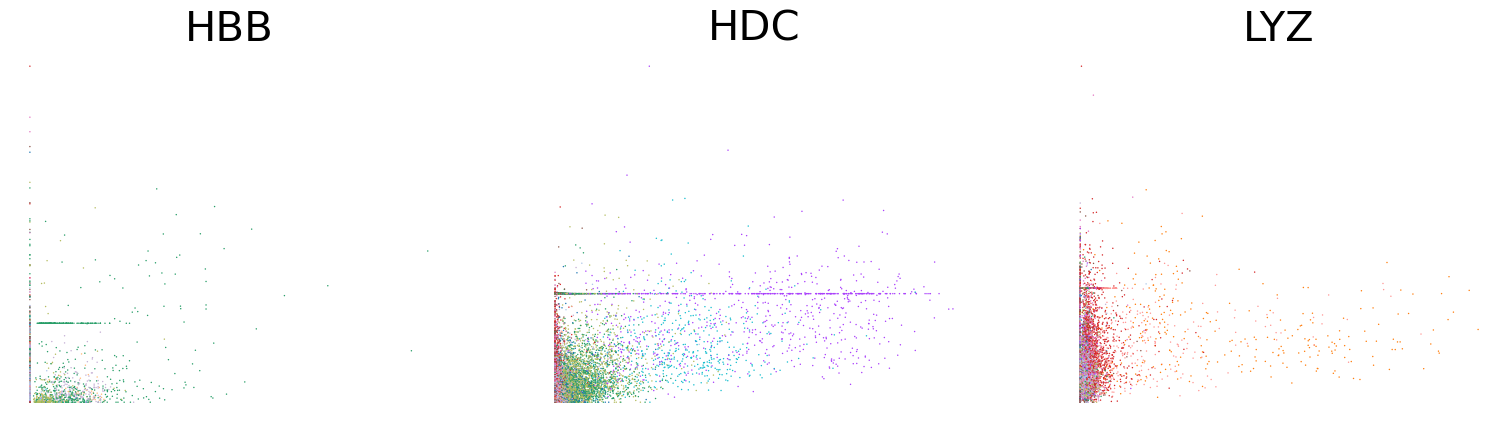
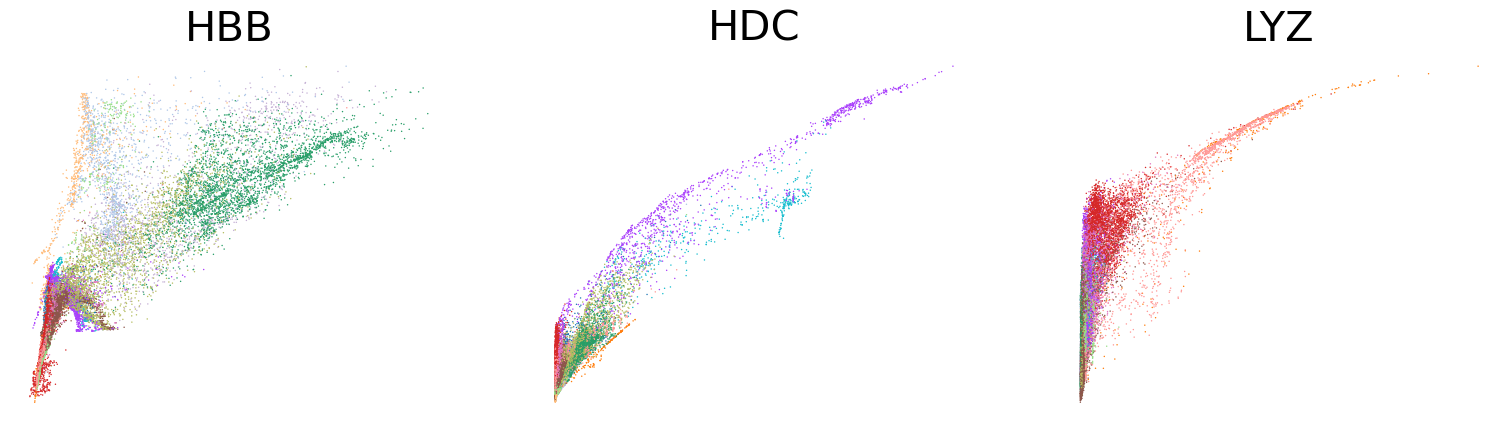
[31]:
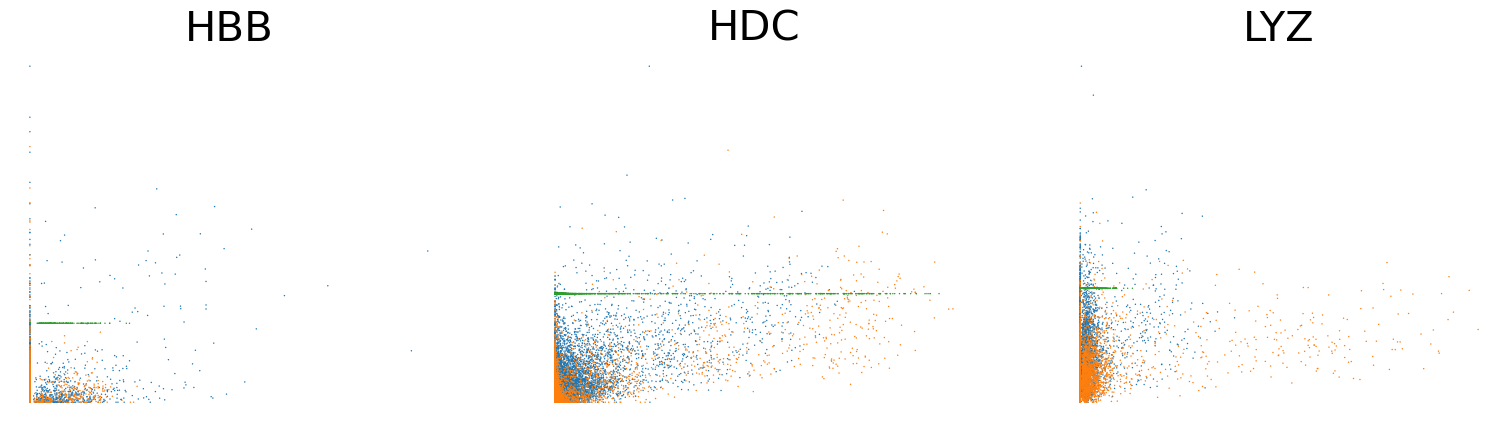
scv.pl.scatter(adata, basis=['HBB', 'HDC', 'LYZ'], x='Mu', y='Mc', color='batch', ncols=3, frameon=False, fontsize=30)
scv.pl.scatter(adata, basis=['HBB', 'HDC', 'LYZ'], x=f'{key}_uhat', y=f'{key}_chat', color='batch', ncols=3, frameon=False, fontsize=30)
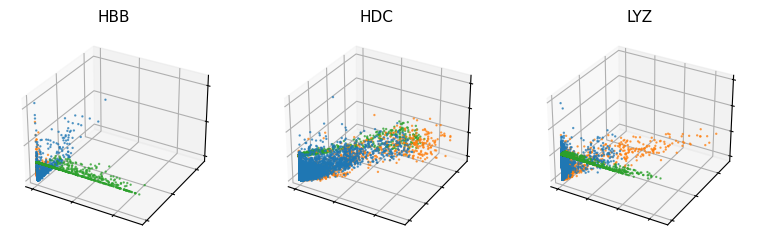
[32]:
vv.scatter_plot(adata, adata_atac, genes=['HBB', 'HDC', 'LYZ'], by='cus', modalities_pred=['Mc', 'Mu', 'Ms'], color_by='batch', n_cols=3, show_pred_only=True, frame_on=True, axis_on=False, pointsize=0.5)
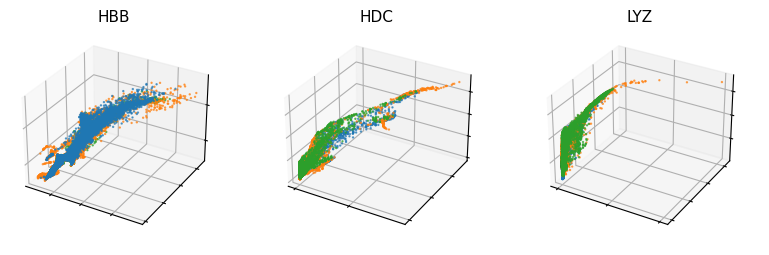
[33]:
vv.scatter_plot(adata, adata_atac, genes=['HBB', 'HDC', 'LYZ'], by='cus', modalities=[f'{key}_chat', f'{key}_uhat', f'{key}_shat'], color_by='batch', n_cols=3, show_pred_only=True, frame_on=True, axis_on=False, pointsize=0.5)
[34]:
# Phase portraits of genes, original, reconstructed and batch-corrected, and reconstructed and not-batch-corrected
scv.pl.scatter(adata, basis=top_genes[:20], color='leiden', ncols=5)
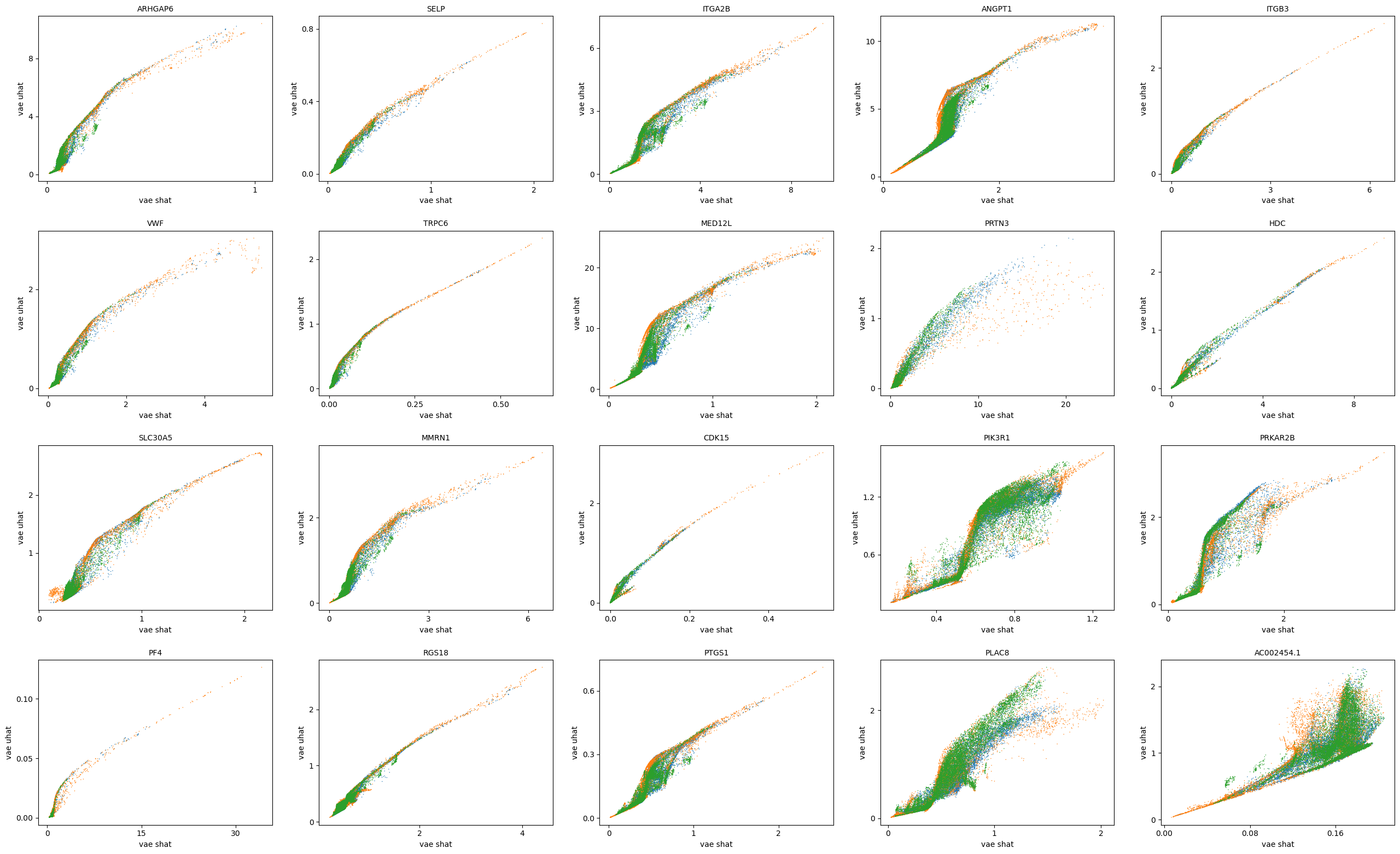
scv.pl.scatter(adata, basis=top_genes[:20], x=f'{key}_shat', y=f'{key}_uhat', color='leiden', ncols=5)
scv.pl.scatter(adata, basis=top_genes[:20], x=f'{key}_shat_batch', y=f'{key}_uhat_batch', color='leiden', ncols=5)
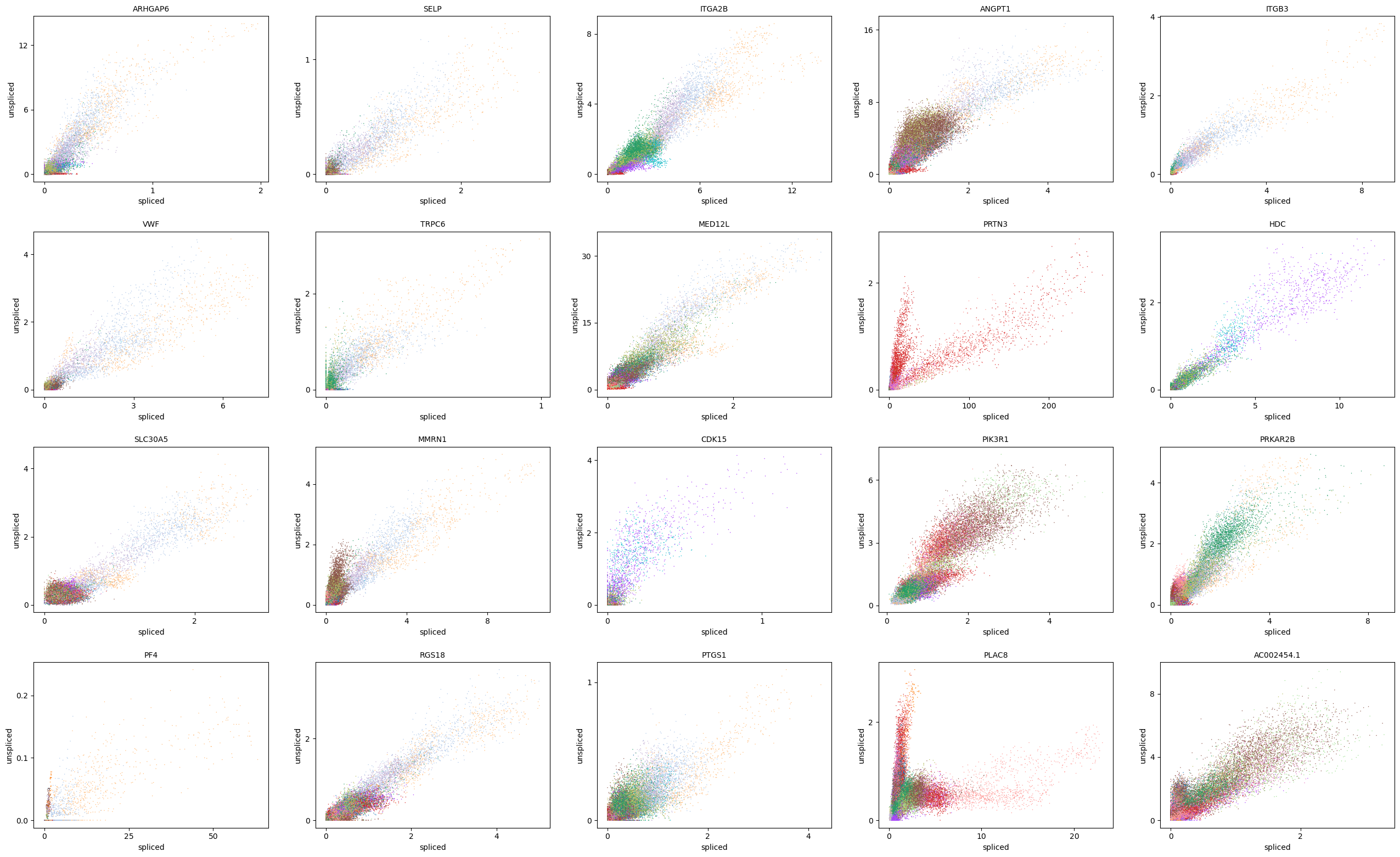
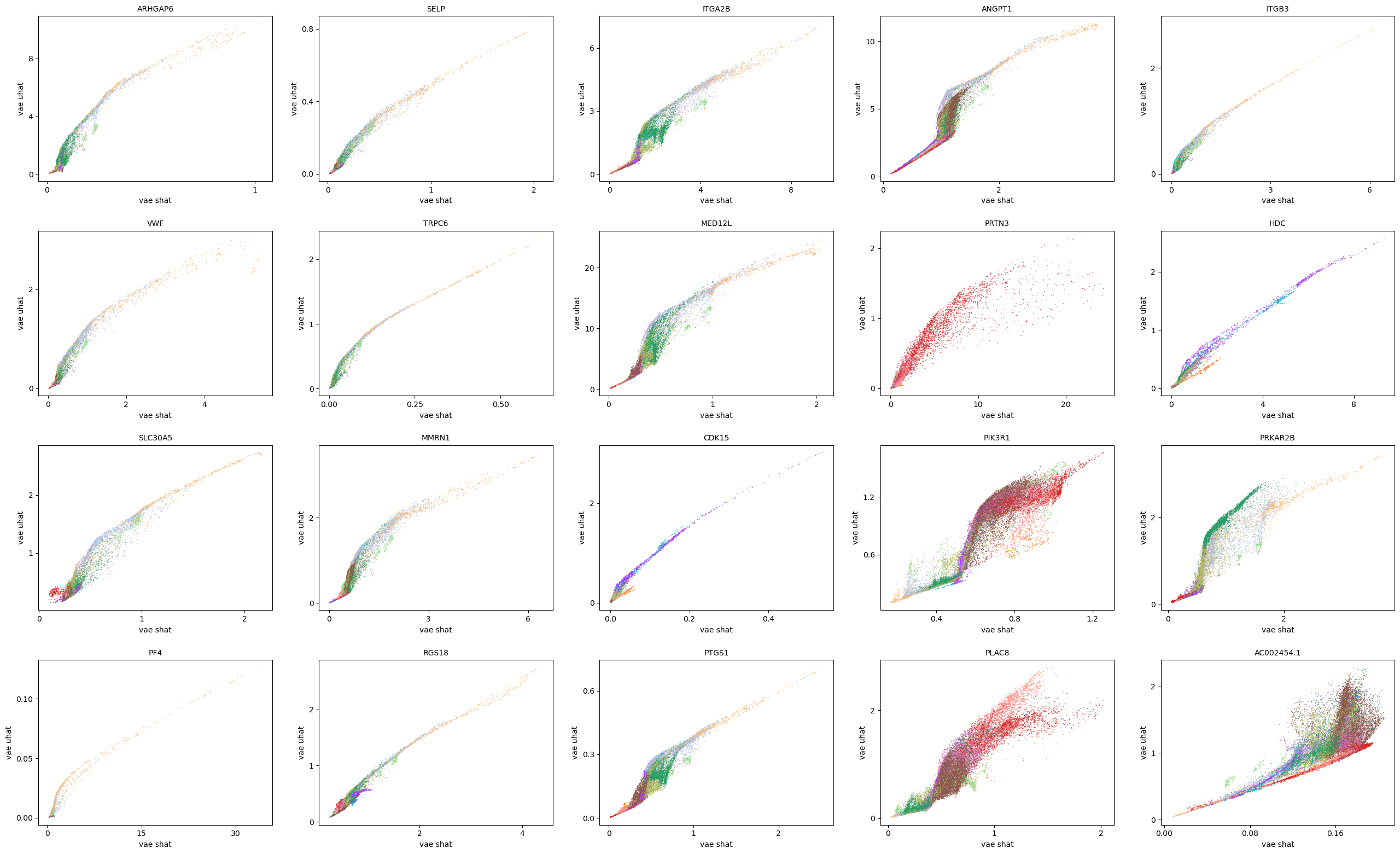
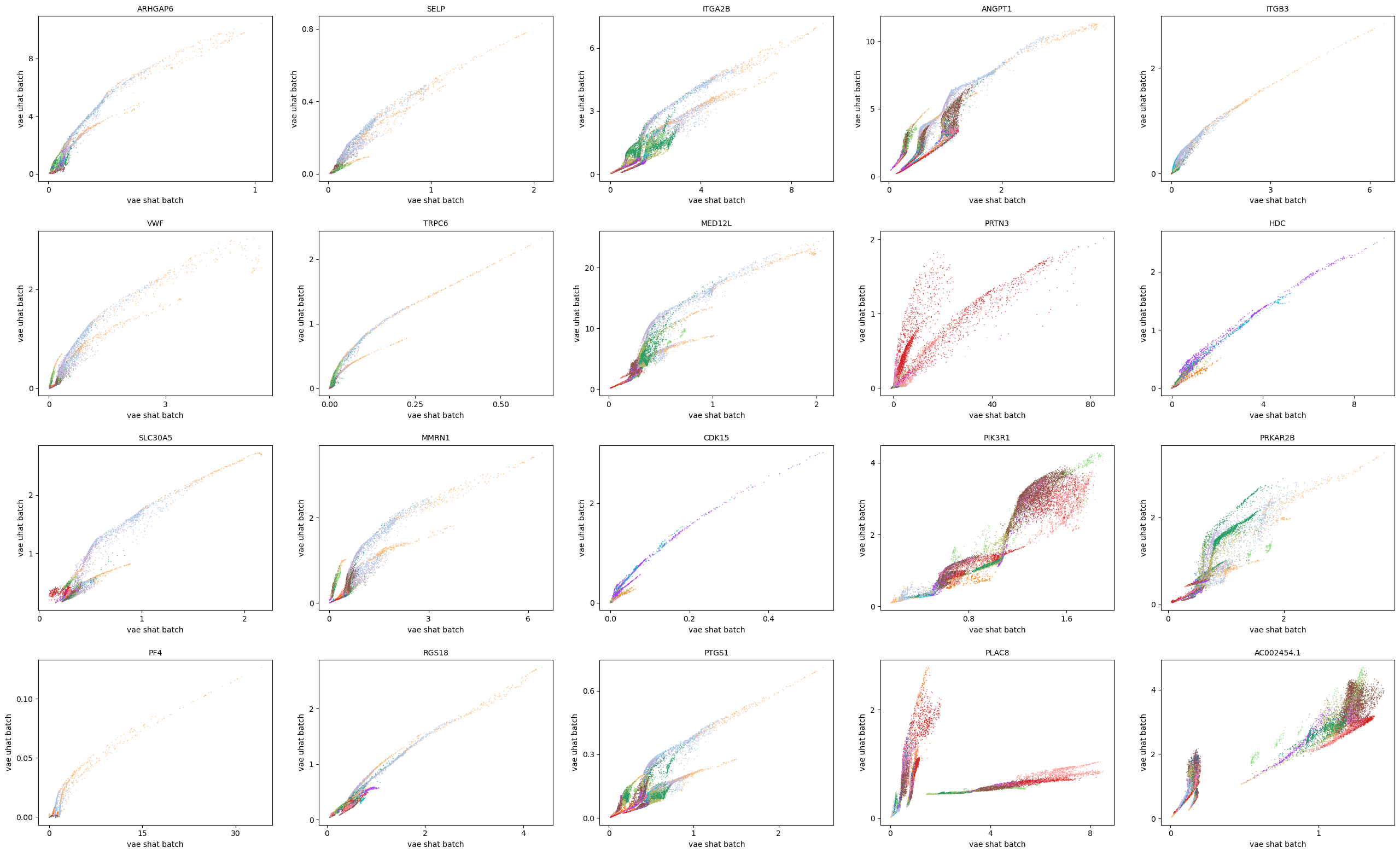
[35]:
scv.pl.scatter(adata, basis=top_genes[:20], color='batch', ncols=5)
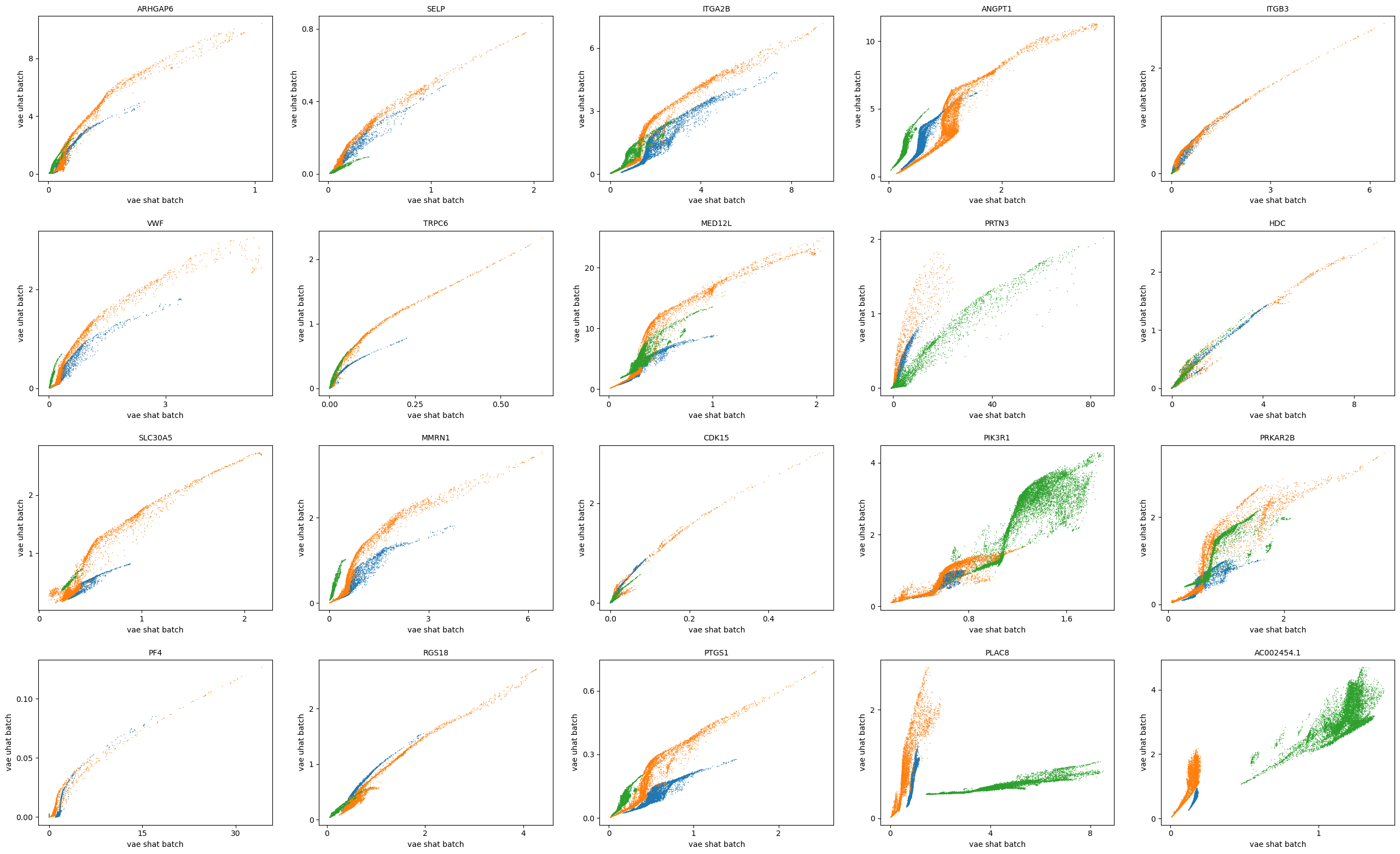
scv.pl.scatter(adata, basis=top_genes[:20], x=f'{key}_shat', y=f'{key}_uhat', color='batch', ncols=5)
scv.pl.scatter(adata, basis=top_genes[:20], x=f'{key}_shat_batch', y=f'{key}_uhat_batch', color='batch', ncols=5)
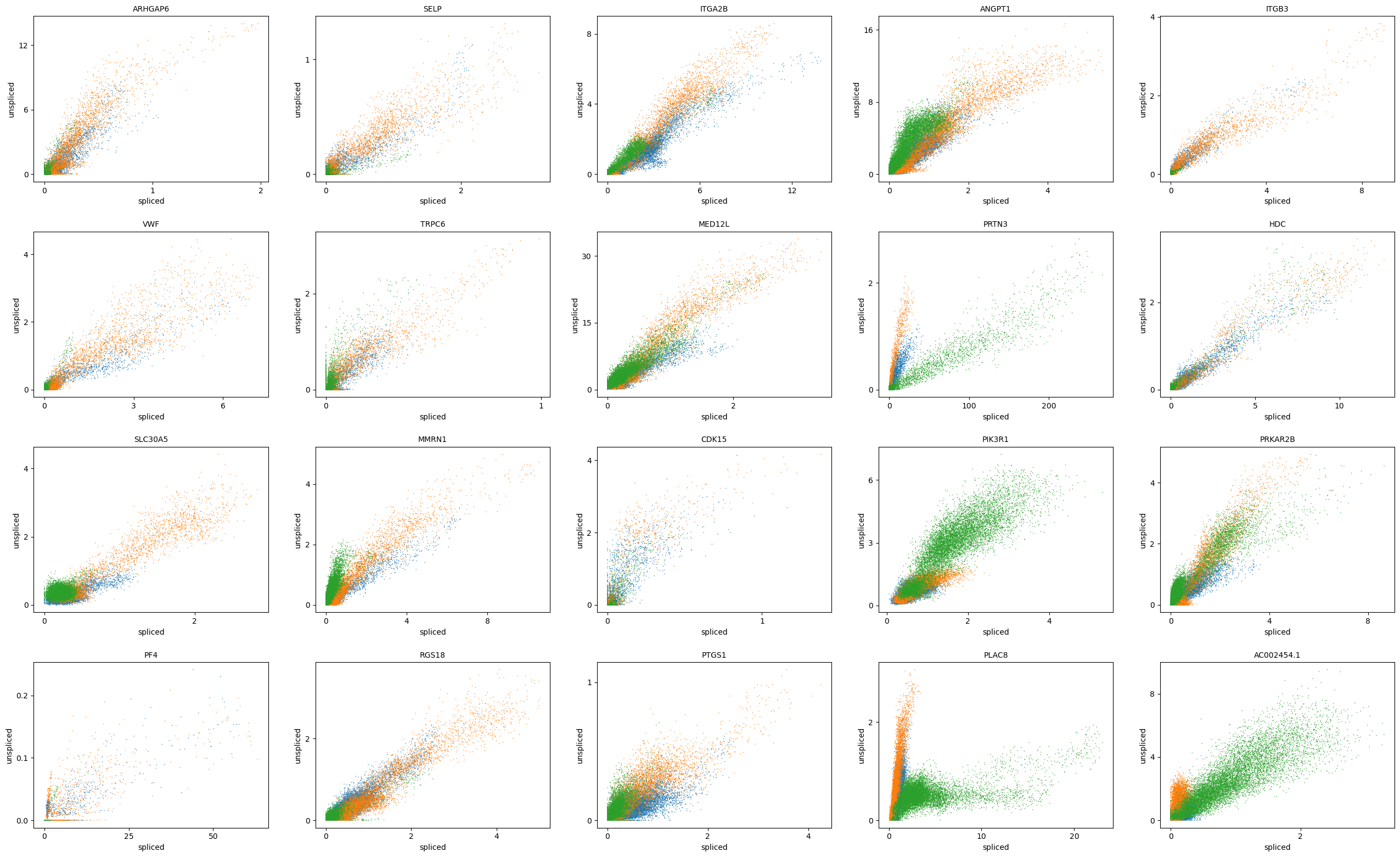
In silico perturbation of PU.1 and GATA1
[38]:
'SPI1' in adata.var_names, 'GATA1' in adata.var_names
[38]:
(True, True)
[39]:
scv.pl.scatter(adata, color='SPI1', basis='z_umap', size=10, title='')
scv.pl.scatter(adata, color='GATA1', basis='z_umap', size=10, title='')

[40]:
c1 = adata_atac.layers['Mc'].copy()
u1 = adata.layers['Mu'].copy()
s1 = adata.layers['Ms'].copy()
c2 = adata_atac.layers['Mc'].copy()
u2 = adata.layers['Mu'].copy()
s2 = adata.layers['Ms'].copy()
[41]:
c1[:, np.where(adata.var_names == 'SPI1')[0][0]] = 0
u1[:, np.where(adata.var_names == 'SPI1')[0][0]] = 0
s1[:, np.where(adata.var_names == 'SPI1')[0][0]] = 0
c2[:, np.where(adata.var_names == 'GATA1')[0][0]] = 0
u2[:, np.where(adata.var_names == 'GATA1')[0][0]] = 0
s2[:, np.where(adata.var_names == 'GATA1')[0][0]] = 0
[42]:
spi1_ko_set = model.prepare_dataset(c1, u1, s1, only_full=True)
gata1_ko_set = model.prepare_dataset(c2, u2, s2, only_full=True)
[43]:
# Get PU.1 KO results
out1 = model.test(spi1_ko_set, covar=model.var_to_regress.detach().cpu().numpy())
chat1, uhat1, shat1, _, _, _, z1, std_z1, t1, std_t1, t_1, kc1, rho1 = out1
Using reference batch for latent variable computation.
[44]:
adata_spi1 = ad.AnnData(np.zeros((adata.n_obs, adata.n_vars*3)), obs=adata.obs)
adata_spi1.uns = adata.uns
adata_spi1.obsm = adata.obsm
adata_spi1.obsp = adata.obsp
[45]:
norm_c = np.linalg.norm(adata.layers[f'{key}_chat'][:,adata.var[f'{key}_velocity_genes'].values], axis=1).reshape(-1, 1) + 1e-10
norm_u = np.linalg.norm(adata.layers[f'{key}_uhat'][:,adata.var[f'{key}_velocity_genes'].values], axis=1).reshape(-1, 1) + 1e-10
norm_s = np.linalg.norm(adata.layers[f'{key}_shat'][:,adata.var[f'{key}_velocity_genes'].values], axis=1).reshape(-1, 1) + 1e-10
[46]:
adata_spi1.layers['cus'] = np.concatenate((adata.layers[f'{key}_chat']/norm_c, adata.layers[f'{key}_uhat']/norm_u, adata.layers[f'{key}_shat']/norm_s), 1)
adata_spi1.layers['spi1_velocity'] = np.concatenate((chat1/norm_c, uhat1/norm_u, shat1/norm_s), 1) - adata_spi1.layers['cus']
adata_spi1.var['spi1_velocity_genes'] = np.concatenate((adata.var[f'{key}_velocity_genes'].values, adata.var[f'{key}_velocity_genes'].values, adata.var[f'{key}_velocity_genes'].values))
adata_spi1.uns["spi1_velocity_params"] = {'mode': 'dynamical'}
adata_spi1.obsm['X_z_umap'] = z_umap
vv.model.velocity_graph(adata_spi1, key='spi1', xkey='cus', batch_corrected=True)
computing velocity graph (using 1/8 cores)
finished (0:03:39) --> added
'spi1_velocity_norm_graph', sparse matrix with cosine correlations (adata.uns)
[47]:
# Plot perturbation force
vv.velocity_embedding_stream(adata_spi1, key='spi1', basis='z_umap', color='leiden', title="", figsize=(8,6), legend_loc='none')
vv.velocity_embedding_stream(adata_spi1, key='spi1', basis='z_umap', color='batch', title="", figsize=(8,6), legend_loc='none')
computing velocity embedding
finished (0:00:03) --> added
'spi1_velocity_norm_z_umap', embedded velocity vectors (adata.obsm)


[48]:
adata.obs['spi1_ko_time_diff'] = np.abs(t_1 - adata.obs[f'{key}_time'])
[49]:
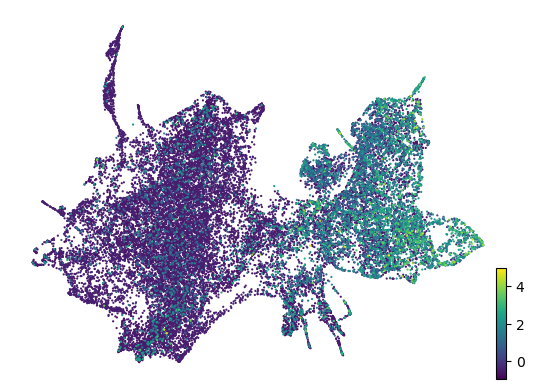
# Plot time shift
scv.pl.scatter(adata, basis='z_umap', color='spi1_ko_time_diff', legend_loc='right', color_map='gnuplot', size=20, title='')
[50]:
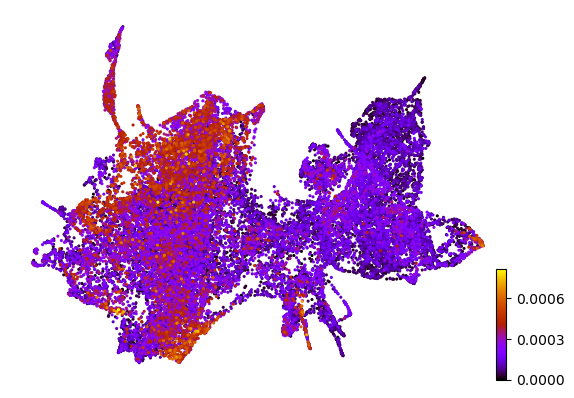
# Plot latent space shift
pca = PCA(n_components=1, random_state=42)
z_pca = pca.fit_transform(np.abs(z1-adata.obsm[f'{key}_z']))
adata.obs['z_pca_spi1_ko'] = z_pca
scv.pl.scatter(adata, color='z_pca_spi1_ko', basis='z_umap', size=10, title='')

[51]:
# Get GATA1 KO results
out2 = model.test(gata1_ko_set, covar=model.var_to_regress.detach().cpu().numpy())
chat2, uhat2, shat2, _, _, _, z2, std_z2, t2, std_t2, t_2, kc2, rho2 = out2
Using reference batch for latent variable computation.
[52]:
adata_gata1 = ad.AnnData(np.zeros((adata.n_obs, adata.n_vars*3)), obs=adata.obs)
adata_gata1.uns = adata.uns
adata_gata1.obsm = adata.obsm
adata_gata1.obsp = adata.obsp
[53]:
adata_gata1.layers['cus'] = np.concatenate((adata.layers[f'{key}_chat']/norm_c, adata.layers[f'{key}_uhat']/norm_u, adata.layers[f'{key}_shat']/norm_s), 1)
adata_gata1.layers['gata1_velocity'] = np.concatenate((chat2/norm_c, uhat2/norm_u, shat2/norm_s), 1) - adata_gata1.layers['cus']
adata_gata1.var['gata1_velocity_genes'] = np.concatenate((adata.var[f'{key}_velocity_genes'].values, adata.var[f'{key}_velocity_genes'].values, adata.var[f'{key}_velocity_genes'].values))
adata_gata1.uns["gata1_velocity_params"] = {'mode': 'dynamical'}
adata_gata1.obsm['X_z_umap'] = z_umap
vv.model.velocity_graph(adata_gata1, key='gata1', xkey='cus', batch_corrected=True)
computing velocity graph (using 1/8 cores)
finished (0:03:42) --> added
'gata1_velocity_norm_graph', sparse matrix with cosine correlations (adata.uns)
[54]:
# Plot perturbation force
vv.velocity_embedding_stream(adata_gata1, key='gata1', basis='z_umap', color='leiden', title="", figsize=(8,6), legend_loc='none')
vv.velocity_embedding_stream(adata_gata1, key='gata1', basis='z_umap', color='batch', title="", figsize=(8,6), legend_loc='none')
computing velocity embedding
finished (0:00:03) --> added
'gata1_velocity_norm_z_umap', embedded velocity vectors (adata.obsm)


[55]:
adata.obs['gata1_ko_time_diff'] = np.abs(t_2 - adata.obs[f'{key}_time'])
[56]:
# Plot time shift
scv.pl.scatter(adata, basis='z_umap', color='gata1_ko_time_diff', legend_loc='right', color_map='gnuplot', size=20, title='')

[57]:
# Plot latent space shift
pca = PCA(n_components=1, random_state=42)
z_pca = pca.fit_transform(np.abs(z2-adata.obsm[f'{key}_z']))
adata.obs['z_pca_gata1_ko'] = z_pca
scv.pl.scatter(adata, color='z_pca_gata1_ko', basis='z_umap', size=10, title='', perc=[0,99.9])
Save the final result
[58]:
adata.write_h5ad(data_path_base+"/final.h5ad")
[ ]: